
Theoretical and Computational Chemistry
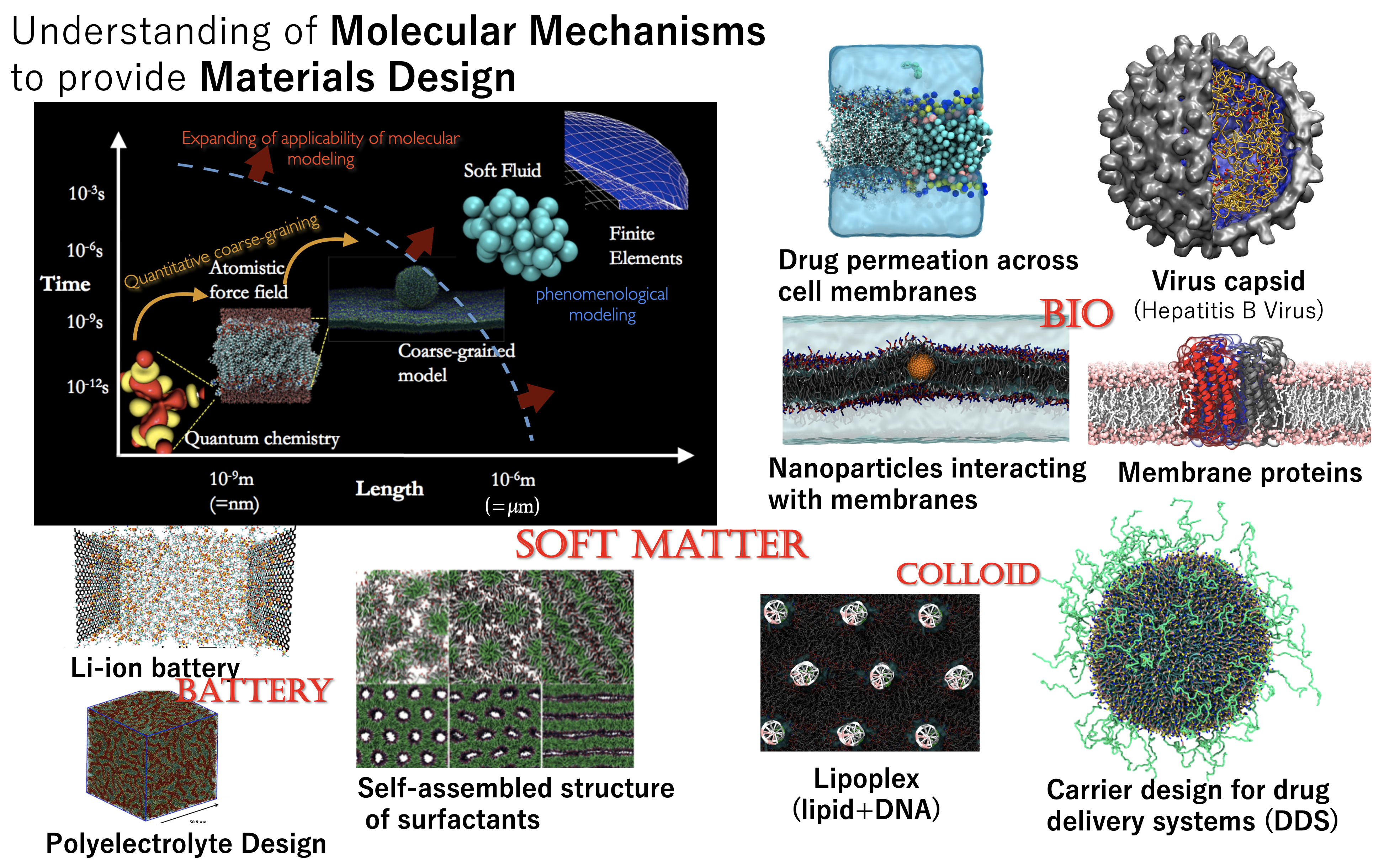
We use molecular simulation techniques to understand and predict various functions of molecules and molecular assemblies such as lipid membranes, aiming to inform the design of new materials. In order to tackle technical issues in bio- and soft-materials, we utilize multiscale molecular modeling, ranging from sub-atomic scales of quantum mechanics, to the coarse-grained molecular level, reaching to the micron scale. By developing these simulation techniques and applying them to contemporary problems, we expand the frontiers of molecular simulations.
 |
|
|---|

To overcome the limitations of molecular simulation, we have developed a quantitative coarse-grained molecular force field, SPICA, to analyze large-scale molecular assembly systems extending into the mesoscale regime. We are now able to treat complex molecular mixtures of submicron size.